nf-core/hlatyping
Precision HLA typing from next-generation sequencing data
Introduction
The hlatyping pipeline can currently deal with two input formats: .fastq{.gz} or .bam. If the input file type is bam, than the pipeline extracts all reads from it and performs an mapping additional step with the yara mapper against the HLA reference sequence. Indices are generated using yara. OptiType uses razers3, which is very memory consuming. In order to avoid memory issues during pipeline execution, we reduce the mapping information on the relevant HLA regions on chromosome 6.
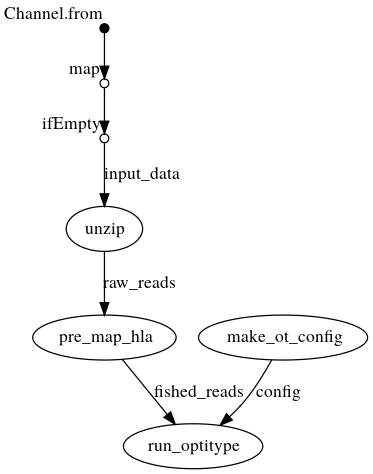
DAG with .fastq{.gz} as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the fastqs are unzipped if they are passed as archives. OptiType is then used for the HLA typing.
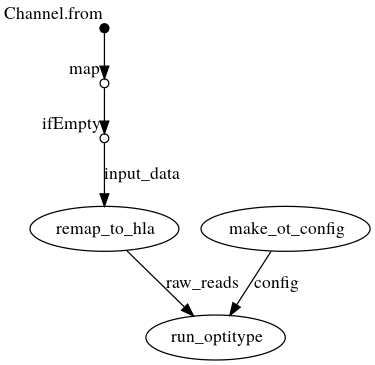
DAG with .bam as input
Creates a config file from the command line arguments, which is then passed to OptiType. In parallel, the reads are extracted from the bam file and mapped again against the HLA reference sequence on chromosome 6. OptiType is then used for the HLA typing.
Samplesheet input
You will need to create a samplesheet with information about the samples you would like to analyse before running the pipeline. Use this parameter to specify its location. It has to be a comma-separated file with 4 columns, and a header row as shown in the examples below.
--input '[path to samplesheet file]'Multiple runs
The sample identifiers have to be specified with the fastq files and the sequencing type:
sample,fastq_1,fastq_2,seq_type
CONTROL_PE,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,dna
CONTROL_SE,AEG588A1_S1_L003_R1_001.fastq.gz,AEG588A1_S1_L003_R2_001.fastq.gz,dnaFull samplesheet
The pipeline will auto-detect whether a sample is single- or paired-end using the information provided in the samplesheet. The samplesheet can have as many columns as you desire, however, there is a strict requirement for the first 4 columns to match those defined in the table below.
A final samplesheet file consisting of both single- and paired-end data may look something like the one below.
sample,fastq_1,fastq_2,seq_type
CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,dna
CONTROL_REP2,AEG588A2_S2_L002_R1_001.fastq.gz,AEG588A2_S2_L002_R2_001.fastq.gz,dna
CONTROL_REP3,AEG588A3_S3_L002_R1_001.fastq.gz,AEG588A3_S3_L002_R2_001.fastq.gz,dna
TREATMENT_REP1,AEG588A4_S4_L003_R1_001.fastq.gz,,dna
TREATMENT_REP2,AEG588A5_S5_L003_R1_001.fastq.gz,,dna
TREATMENT_REP3,AEG588A6_S6_L003_R1_001.fastq.gz,,dna
TREATMENT_REP3,AEG588A6_S6_L004_R1_001.fastq.gz,,dnaThe pipeline can also process BAM files. If you want to process a BAM file, just add the corresponding column to the samplesheet and provide the full path to the file. FASTQ and BAM files can be mixed in the same sample sheet.
sample,fastq_1,fastq_2,bam,seq_type
CONTROL_REP1,AEG588A1_S1_L002_R1_001.fastq.gz,AEG588A1_S1_L002_R2_001.fastq.gz,,dna
CONTROL_REP2,AEG588A2_S2_L002_R1_001.fastq.gz,AEG588A2_S2_L002_R2_001.fastq.gz,,rna
TREATMENT_REP1,AEG588A4_S4_L003_R1_001.bam,,dna| Column | Description |
|---|---|
sample | Custom sample name. |
fastq_1 | Full path to FastQ file for Illumina short reads 1. File has to be gzipped and have the extension “.fastq.gz” or “.fq.gz”. |
fastq_2 | Full path to FastQ file for Illumina short reads 2. File has to be gzipped and have the extension “.fastq.gz” or “.fq.gz”. |
bam | OPTIONAL. Full path to BAM file. |
seq_type | DNA or RNA. |
An example samplesheet has been provided with the pipeline.
Running the pipeline
The typical command for running the pipeline is as follows:
nextflow run nf-core/hlatyping --input samplesheet.csv --outdir <OUTDIR> --genome GRCh37 -profile dockerThis will launch the pipeline with the docker configuration profile. See below for more information about profiles.
Note that the pipeline will create the following files in your working directory:
work # Directory containing the nextflow working files
<OUTDIR> # Finished results in specified location (defined with --outdir)
.nextflow_log # Log file from Nextflow
# Other nextflow hidden files, eg. history of pipeline runs and old logs.HLA references
The nf-core/hlatyping pipeline uses a default HLA reference which is located in the pipelines root directory in ./data/references. The references are based on the IMGT/HLA Release 3.14.0, July 2013, and have been processed as described in the publication of OptiType. The reference is automatically set during the pipeline execution based on the information provided in the seq_type column of the samplesheet (dna or rna).
You can always download new versions from the HLA database, but be aware that these allele sets are missing intron sequence information, which will have a negative influence in the HLA typing outcome in case of DNAseq.
We are currently looking into a dynamic solution, in order to build pre-processed input HLA references from current HLA allele information from the IPD-IMGT/HLA database.
Updating the pipeline
When you run the above command, Nextflow automatically pulls the pipeline code from GitHub and stores it as a cached version. When running the pipeline after this, it will always use the cached version if available - even if the pipeline has been updated since. To make sure that you’re running the latest version of the pipeline, make sure that you regularly update the cached version of the pipeline:
nextflow pull nf-core/hlatypingReproducibility
It is a good idea to specify a pipeline version when running the pipeline on your data. This ensures that a specific version of the pipeline code and software are used when you run your pipeline. If you keep using the same tag, you’ll be running the same version of the pipeline, even if there have been changes to the code since.
First, go to the nf-core/hlatyping releases page and find the latest version number - numeric only (eg. 1.3.1). Then specify this when running the pipeline with -r (one hyphen) - eg. -r 1.3.1.
This version number will be logged in reports when you run the pipeline, so that you’ll know what you used when you look back in the future.
Core Nextflow arguments
NB: These options are part of Nextflow and use a single hyphen (pipeline parameters use a double-hyphen).
-profile
Use this parameter to choose a configuration profile. Profiles can give configuration presets for different compute environments.
Several generic profiles are bundled with the pipeline which instruct the pipeline to use software packaged using different methods (Docker, Singularity, Podman, Shifter, Charliecloud, Conda) - see below. When using Biocontainers, most of these software packaging methods pull Docker containers from quay.io e.g FastQC except for Singularity which directly downloads Singularity images via https hosted by the Galaxy project and Conda which downloads and installs software locally from Bioconda.
We highly recommend the use of Docker or Singularity containers for full pipeline reproducibility, however when this is not possible, Conda is also supported.
The pipeline also dynamically loads configurations from https://github.com/nf-core/configs when it runs, making multiple config profiles for various institutional clusters available at run time. For more information and to see if your system is available in these configs please see the nf-core/configs documentation.
Note that multiple profiles can be loaded, for example: -profile test,docker - the order of arguments is important!
They are loaded in sequence, so later profiles can overwrite earlier profiles.
If -profile is not specified, the pipeline will run locally and expect all software to be installed and available on the PATH. This is not recommended.
docker- A generic configuration profile to be used with Docker
singularity- A generic configuration profile to be used with Singularity
podman- A generic configuration profile to be used with Podman
shifter- A generic configuration profile to be used with Shifter
charliecloud- A generic configuration profile to be used with Charliecloud
conda- A generic configuration profile to be used with Conda. Please only use Conda as a last resort i.e. when it’s not possible to run the pipeline with Docker, Singularity, Podman, Shifter or Charliecloud.
test- A profile with a complete configuration for automated testing
- Includes links to test data so needs no other parameters
-resume
Specify this when restarting a pipeline. Nextflow will use cached results from any pipeline steps where the inputs are the same, continuing from where it got to previously. For input to be considered the same, not only the names must be identical but the files’ contents as well. For more info about this parameter, see this blog post.
You can also supply a run name to resume a specific run: -resume [run-name]. Use the nextflow log command to show previous run names.
-c
Specify the path to a specific config file (this is a core Nextflow command). See the nf-core website documentation for more information.
Custom configuration
Resource requests
Whilst the default requirements set within the pipeline will hopefully work for most people and with most input data, you may find that you want to customise the compute resources that the pipeline requests. Each step in the pipeline has a default set of requirements for number of CPUs, memory and time. For most of the steps in the pipeline, if the job exits with any of the error codes specified here it will automatically be resubmitted with higher requests (2 x original, then 3 x original). If it still fails after the third attempt then the pipeline execution is stopped.
For example, if the nf-core/rnaseq pipeline is failing after multiple re-submissions of the STAR_ALIGN process due to an exit code of 137 this would indicate that there is an out of memory issue:
[62/149eb0] NOTE: Process `NFCORE_RNASEQ:RNASEQ:ALIGN_STAR:STAR_ALIGN (WT_REP1)` terminated with an error exit status (137) -- Execution is retried (1)
Error executing process > 'NFCORE_RNASEQ:RNASEQ:ALIGN_STAR:STAR_ALIGN (WT_REP1)'
Caused by:
Process `NFCORE_RNASEQ:RNASEQ:ALIGN_STAR:STAR_ALIGN (WT_REP1)` terminated with an error exit status (137)
Command executed:
STAR \
--genomeDir star \
--readFilesIn WT_REP1_trimmed.fq.gz \
--runThreadN 2 \
--outFileNamePrefix WT_REP1. \
<TRUNCATED>
Command exit status:
137
Command output:
(empty)
Command error:
.command.sh: line 9: 30 Killed STAR --genomeDir star --readFilesIn WT_REP1_trimmed.fq.gz --runThreadN 2 --outFileNamePrefix WT_REP1. <TRUNCATED>
Work dir:
/home/pipelinetest/work/9d/172ca5881234073e8d76f2a19c88fb
Tip: you can replicate the issue by changing to the process work dir and entering the command `bash .command.run`To bypass this error you would need to find exactly which resources are set by the STAR_ALIGN process. The quickest way is to search for process STAR_ALIGN in the nf-core/rnaseq Github repo.
We have standardised the structure of Nextflow DSL2 pipelines such that all module files will be present in the modules/ directory and so, based on the search results, the file we want is modules/nf-core/software/star/align/main.nf.
If you click on the link to that file you will notice that there is a label directive at the top of the module that is set to label process_high.
The Nextflow label directive allows us to organise workflow processes in separate groups which can be referenced in a configuration file to select and configure subset of processes having similar computing requirements.
The default values for the process_high label are set in the pipeline’s base.config which in this case is defined as 72GB.
Providing you haven’t set any other standard nf-core parameters to cap the maximum resources used by the pipeline then we can try and bypass the STAR_ALIGN process failure by creating a custom config file that sets at least 72GB of memory, in this case increased to 100GB.
The custom config below can then be provided to the pipeline via the -c parameter as highlighted in previous sections.
process {
withName: 'NFCORE_RNASEQ:RNASEQ:ALIGN_STAR:STAR_ALIGN' {
memory = 100.GB
}
}NB: We specify the full process name i.e.
NFCORE_RNASEQ:RNASEQ:ALIGN_STAR:STAR_ALIGNin the config file because this takes priority over the short name (STAR_ALIGN) and allows existing configuration using the full process name to be correctly overridden.If you get a warning suggesting that the process selector isn’t recognised check that the process name has been specified correctly.
Updating containers
The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. If for some reason you need to use a different version of a particular tool with the pipeline then you just need to identify the process name and override the Nextflow container definition for that process using the withName declaration. For example, in the nf-core/viralrecon pipeline a tool called Pangolin has been used during the COVID-19 pandemic to assign lineages to SARS-CoV-2 genome sequenced samples. Given that the lineage assignments change quite frequently it doesn’t make sense to re-release the nf-core/viralrecon everytime a new version of Pangolin has been released. However, you can override the default container used by the pipeline by creating a custom config file and passing it as a command-line argument via -c custom.config.
-
Check the default version used by the pipeline in the module file for Pangolin
-
Find the latest version of the Biocontainer available on Quay.io
-
Create the custom config accordingly:
-
For Docker:
process { withName: PANGOLIN { container = 'quay.io/biocontainers/pangolin:3.0.5--pyhdfd78af_0' } } -
For Singularity:
process { withName: PANGOLIN { container = 'https://depot.galaxyproject.org/singularity/pangolin:3.0.5--pyhdfd78af_0' } } -
For Conda:
process { withName: PANGOLIN { conda = 'bioconda::pangolin=3.0.5' } }
-
NB: If you wish to periodically update individual tool-specific results (e.g. Pangolin) generated by the pipeline then you must ensure to keep the
work/directory otherwise the-resumeability of the pipeline will be compromised and it will restart from scratch.
nf-core/configs
In most cases, you will only need to create a custom config as a one-off but if you and others within your organisation are likely to be running nf-core pipelines regularly and need to use the same settings regularly it may be a good idea to request that your custom config file is uploaded to the nf-core/configs git repository. Before you do this please can you test that the config file works with your pipeline of choice using the -c parameter. You can then create a pull request to the nf-core/configs repository with the addition of your config file, associated documentation file (see examples in nf-core/configs/docs), and amending nfcore_custom.config to include your custom profile.
See the main Nextflow documentation for more information about creating your own configuration files.
If you have any questions or issues please send us a message on Slack on the #configs channel.
Azure Resource Requests
To be used with the azurebatch profile by specifying the -profile azurebatch.
We recommend providing a compute params.vm_type of Standard_D16_v3 VMs by default but these options can be changed if required.
Note that the choice of VM size depends on your quota and the overall workload during the analysis. For a thorough list, please refer the Azure Sizes for virtual machines in Azure.
Running in the background
Nextflow handles job submissions and supervises the running jobs. The Nextflow process must run until the pipeline is finished.
The Nextflow -bg flag launches Nextflow in the background, detached from your terminal so that the workflow does not stop if you log out of your session. The logs are saved to a file.
Alternatively, you can use screen / tmux or similar tool to create a detached session which you can log back into at a later time.
Some HPC setups also allow you to run nextflow within a cluster job submitted your job scheduler (from where it submits more jobs).
Nextflow memory requirements
In some cases, the Nextflow Java virtual machines can start to request a large amount of memory.
We recommend adding the following line to your environment to limit this (typically in ~/.bashrc or ~./bash_profile):
NXF_OPTS='-Xms1g -Xmx4g'